Les fichiers d'entrée/sortie
Vue d'ensemble
VASP nécessite plusiers fichiers d'entrée avec un nom et un format imposés. Ils doivent se trouver dans le dossier d'exécution du code. Plusieurs fichiers de sortie sont créés durant l'exécution du code et se trouvent également dans le dossier d'exécution. Certains fichiers ne sont présents que dans certaines conditions. Voici une schéma général des entrées/sorties avec les principaux fichiers de sortie :
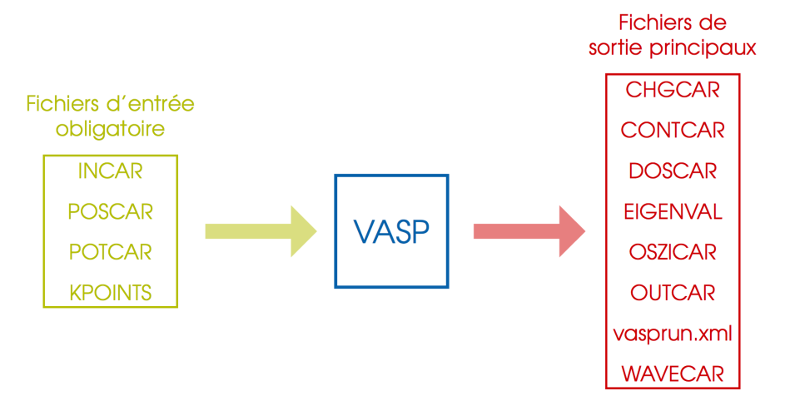
Il est recommandé de créer un dossier pour chaque nouveau calcul, sauf si l'on souhaite écraser les données présentes dans le dossier.
Les fichiers d'entrée
INCAR: Type de calcul et paramètres du calculPOSCAR: Positions initiales des atomesKPOINTS: Grille de points k utiliséePOTCAR: Contient les pseudo-potentiels utilisés
Attention : le nom des fichiers est imposé.
Fichiers de sortie
Parmi les principaux fichiers de sortie, on notera :
OUTCAR: Listing du calcul, informations générales ...OSZICAR: Itérations électroniques et ioniques, utile pour suivre la convergence du calcul.WAVECAR: fichier binaire contenant la fonciton d'onde finale.CHGCAR: densité électronique finale discrétisée sur une grille.CONTCAR: structure finaleDOSCAR: Densité d'étatsEIGENVAL: Valeurs propres de l'hamiltonien pour chaque bande et pour chaque point k.vasprun.xml: Ce fichier contient toutes les informations précédentes, sauf la densité électronique et la fonction d'onde, mais au format xml. Il est utilisé par certains outils de post-traitement, le langage xml rendant sa lecture plus facile que celle du fichierOUTCAR.
Le fichier INCAR
Le fichier INCAR
contient les paramètres du calcul et définit donc le type de simulation.
Les paramètres sont donnés dans une syntaxe de type clef = valeur.
La quasi totalité des mots clefs disponnibles sont présents sur le
wiki des mots clefs.
Dans les exemples suivants :
- la première ligne est une ligne de titre
- les mots clefs sont regroupés lorsqu'ils agissent sur la même partie du calcul
- les lignes sans signe égal sont traitées comme des commentaires et sont facultatives
Simple calcul d'énergie
Dans ce calcul, la structure n'est pas relaxée. Seule la densité électronique est relaxée.
Electronic
PREC = Accurate
EDIFF = 1e-6
NELMIN = 4
ENCUT = 400
LORBIT = 11
Ionic relaxation
NSW = 0
smearing
ISMEAR = -5
On notera des paramètres précisant la précision du calcul (ENCUT, PREC) ou le
seuil de convergence (EDIFF).
Optimisation de la géométrie
Dans le jargon utilisé par VASP, les ions désignent généralement les noyaux des atomes. Ici ionic relaxation se réfère donc à la relaxation de la structure.
Ionic relaxation
Electronic
PREC = Accurate
EDIFF = 1e-6
NELMIN = 4
ENCUT = 400
LORBIT = 11
Ionic relaxation
NSW = 200
ISIF = 2
IBRION = 2
EDIFFG = -0.01
smearing
ISMEAR = 0
SIGMA = 0.05
Les nouveaux paramètres indiquent le nombre d'itérations maximum pour l'optimisation
de la structure (NSW), l'algorithme utilisé (IBRION), les paramètres structuraux
optimisés (ISIF) et la précision attendue (EDIFFG).
Le fichier POSCAR
Le fichier POSCAR contient la structure initiale. Il contient le réseau de bravais et les positions atomiques.
Nous allons voir comment écrire le fichier POSCAR d'une structure antifluorine.
Prennons le cas de dont la structure est représenté ci-dessous.
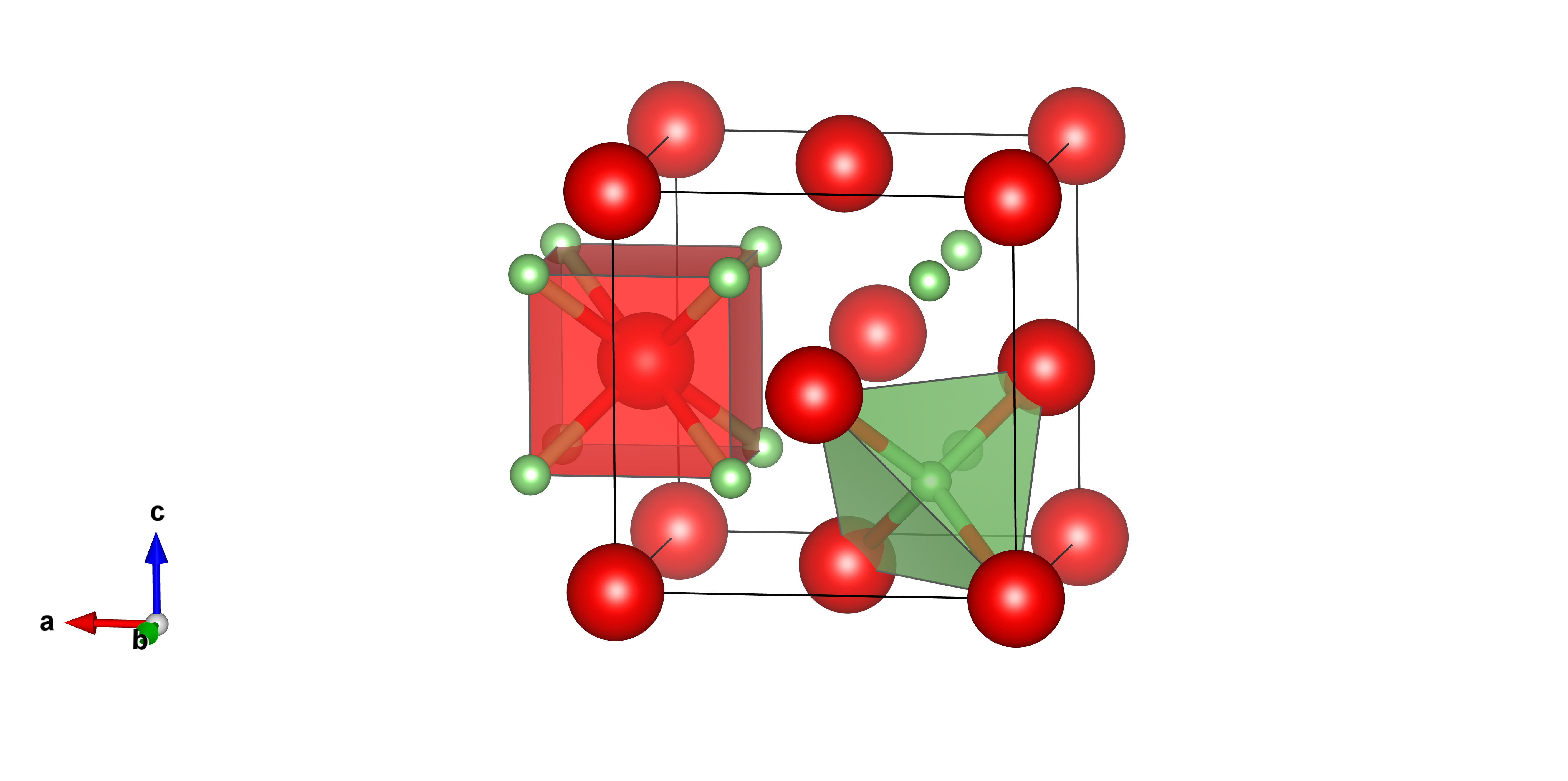
Le fichier POSCAR de cette structure dans sa maille représentative cubique faces
centrées est :
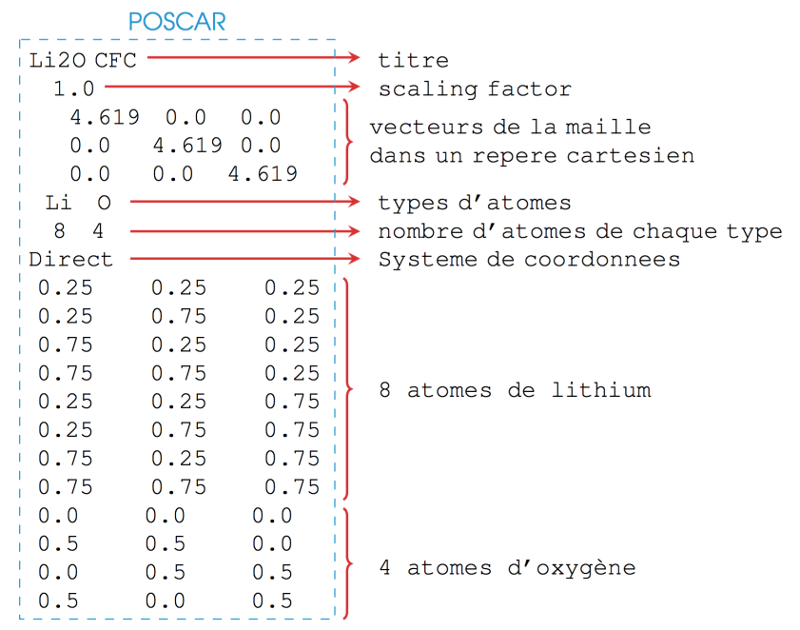
Le fichier CONTCAR est écrit dans un format identique au fichier POSCAR.
Les fichiers POSCAR et CONTCAR peuvent également contenir
les coordonnées des vecteurs vitesses de chaque atome à la suite
des positions atomiques séparées par une ligne blanche.
Le fichier KPOINTS
Le fichier KPOINTS contient la grille de point k utilisée pour intégrer la première zone de Brillouin de l'espace réciproque. C'est la principale différence entre un code périodique comme VASP et un code moléculaire (en dehors de la base).
La grille de points k doit paver la première zone de Brillouin. La taille de la grille
est un compromis entre précision et temps de calcul. Généralement on demande à VASP de
créer la grille automatiquement, le fichier KPOINTS est alors le suivant, pour une
grille centrée sur le point (origine du réseau réciproque) avec 6 points k
suivant chaque direction (216 au total) :
grille automatique
0
Gamma
6 6 6
0 0 0
En réalité, VASP ne fait pas le calcul pour 216 points k. Il cherche ceux qui se trouvent dans la partie irréductible de la première zone de Brillouin et pondère ces points k par le nombre d'homologues qu'ils induisent dans la première zone de Brillouin.
Dans le fichier KPOINTS, on peut également indiquer des lignes de haute symétrie de
l'espace réciproque pour tracer un diagramme de bandes.
Quelques recommandations générales :
- Système non périodique (molécule) : un seul point k, grille 1×1×1
- Le nombre de points k requis est lié aux paramètres de maille. Plus le paramètre de maille est petit plus il faut un grand nombre de points k.
- Pour le calcul d'une DOS il faut un grand nombre de points k.
- Pour une maille hexagonale centrer la grille en .
- Voir le mot clef
KPARpour la parallélisation
Le fichier POTCAR
Le fichier POTCAR contient les pseudos potentiels utilisés pour chaque atome du calcul.
Les fichiers POTCAR sont fournis par VASP et disponnible dans une
base de données.
Pour un élément donné, plusieurs pseudo potentiels peuvent être disponnibles suivant
la fonctionnelle de la densité utilisée pour le construire et le nombre d'électrons
de cœur contenu dans le pseudopotentiel. Voici un aperçu d'un fichier POTCAR et la
descrption de quelques éléments qu'il contient :

Si plusieurs atomes sont présents dans le système on concatène les fichiers POTCAR dans l'ordre
d'apprition des atomes dans le fichier POSCAR.